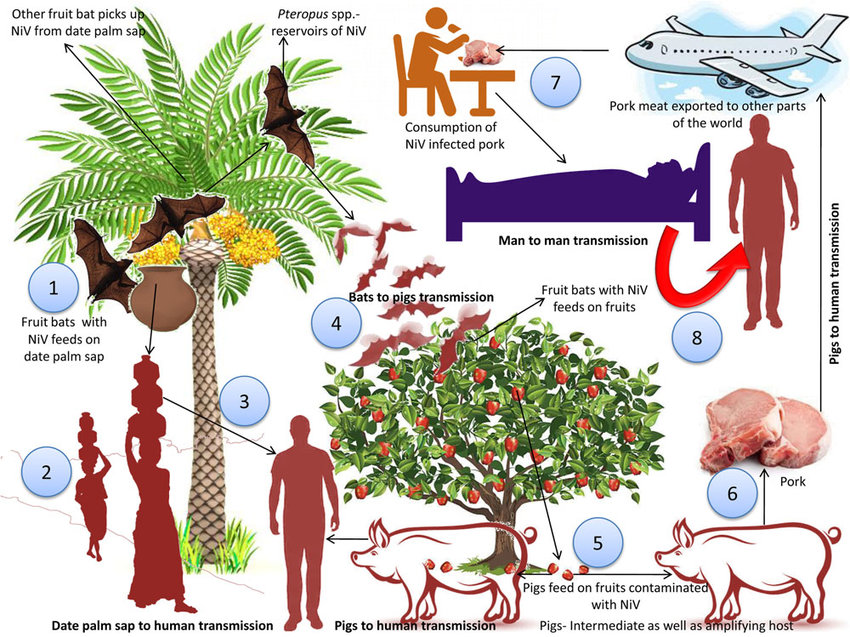
The recent outbreak of covid-19 calls major attention to the threats that zoonotic diseases can pose to human as a species. Out of many hosts and vectors for zoonoses, bats are exceptionally notorious for its ability to transmit diseases to human. This is mainly due to the bat's complex immune system, allowing the mammal to carry pathogens without being affected. This research by Olival et al. focuses on the distribution and mechanisms of Nipah virus, through studying the population genetics of fruit bats.
Nipah virus is a fatal zoonotic RNA paramyxovirus (genus Henipavirus) with bats being its main reservoir host. Depending on the strain, it can have a fatality rate of 40-70% and it has been causing reported deaths continuously since 2001.The goal of this study is to gain better understanding on host population genetic structure in order to improve models of viral circulation dynamics.
Using mitochondrial DNA and nuclear microsatellite markers, the authors were able to measure population structure, demographic history and phylogeography of P. medius in Bangladesh.They also performed a phylogeographic analysis on all known Nipah virus sequences and strains in order to investigate the virus’ evolutionary history.

The results show that P. medius experienced both genetic and morphological differentiation between 2 populations in eastern Bangladesh, producing divergent strains of Nipah virus. The graph above shows that there are several clades of NiV identified in this study. “NiV clades I and II are cocirculating among several Pteropus species (and other bat genera) across South and South‐East Asia, suggesting that NiV is characterized by weak host specificity.” These two NiV clades are shared in P. lylei populations in central Thailand, while only clade II (NiV‐MY) has been found in P. vampyrus and P. hypomelanus. This discovery helps to identify the specific strains with their original hosts, and can be useful to public health measures as different genotypes of NiV can have different clinical presentations in humans and animals.
Demographic analysis also shows that a large population of Pteropus species has existed in Bangladesh since the late Plesitocene, coinciding with the first hominin expansion. This suggests that Nipah virus spillover to human might have a longer history than previously expected.
Olival, K.J., Latinne, A., Islam, A., Epstein, J.H., Hersch, R., Engstrand, R.C., Gurley, E.S., Amato, G., Luby, S.P. and Daszak, P., 2020. Population genetics of fruit bat reservoir informs the dynamics, distribution and diversity of Nipah virus. Molecular Ecology, 29(5), pp.970-985.
Molecular Ecology Journal Update
Monica Cheung– April 23, 2020